Recombinant DNA Technology
Cloning
Discoveries in molecular biology have allowed sci-entists to duplicate natural genetic transfer phenom-ena in the laboratory and to develop methods to in-troduce almost any type of genetic information into an organism. Genetic engineering is the creation of new DNA, usually by linking DNA from different or-ganisms together by artificial means using enzymes known as restriction enzymes. Cloningis the production of many copies of the newly engi-neered DNA. The amplification of a specific cloned gene or genes, cou-pled with a marked increase in production of their protein products, makes it relatively easy to extract and purify these proteins in the labo-ratory.
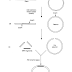
A typical cloning procedure is illustrated in Figure 8-1. A suitable plasmid (vector) is selected in which to insert a desired gene (donor DNA). Both donor DNA and vector are digested with the same restric-tion enzyme, and then incubated together with ligase to join the donor DNA fragments with the plasmid. The result is a recombinant plasmid that contains the desired DNA fragment. The recombinant plasmid is then used to transform a host bacterial cell, creating a new genetic strain of bacteria that stably maintains the recombinant plasmid.


The goal of cloning is to isolate a desired gene or segment of DNA from an organism and introduce it into a suitable host cell to obtain large quantities of the DNA. Often, this donor DNA is used for the large-scale production of important proteins, but the DNA may also be used in the detection of infectious agents or abnormal cells. Normally, the donor DNA is a small portion of the genome of a cell, and it is present as one or two copies in each cell. Therefore, before donor DNA can be extract-ed, a sufficient number of cells containing the desired DNA must be ob-tained, either from a small segment of tissue or by culturing the cells. The cells must then be disrupted and the genetic material (either in chromo-somes or in plasmids) extracted.
Restriction Endonucleases
Restriction endonucleasesare bacterial enzymes that recognize specif-ic nucleotide sequences within a double-stranded DNA molecule and cleave at those locations. These enzymes cut DNA into fragments of var-ious lengths, depending on the number of times the enzyme’s recognition site is repeated within the molecule. Most restriction endonucleases used in cloning recognize base pair sequences four to eight nucleotides long and cut within these sequences. Many restriction enzymes have recogni-tion sequences known as palindromes. Palindromes are sequences that are identical when read in the 5 →3 direction on both strands of the DNA molecule. Restriction endonucleases may cut the DNA to produce fragments with cohesive(“sticky”)endsorblunt ends(see Figure 8-2).
 \
\+cohesive.png "Restriction enzymes may form (a) cohesive or (b) blunt ends")
The resulting fragments of a restriction enzyme digestion can be vi-sualized by a procedure known as electrophoresis. Electrophoresis in-volves the movement of charged molecules or ions through a semisolid support medium under the influence of an electrical field. Agarose gels are common media for the electrophoresis of DNA. The agarose gel, cast as a thin slab in a mold with sample wells at one end, is submerged in a buffer solution with the sample well side toward the negative pole (cath-ode). The samples are dispensed into the wells and a current from the power supply is applied to the system. Since nucleic acids have a nega-tive charge at pH=8.0, they migrate within the gel matrix from the nega-tive to the positive pole (anode) at a rate dependent upon their size, shape, and total charge. DNA molecules are invisible to the naked eye, but can be seen in gels by staining them with a solution of a dye called ethidium bromide, which intercalates between the stacked bases of the DNA mol-ecule and fluoresces.
The graphical representation of recognition sites for two or more re-striction endonucleases is known as a restriction mapfor that molecule. Knowing the restriction maps for commonly used plasmids and bacte-riophage DNA genomes allows scientists to plan cloning stategies for iso-lating and moving around pieces of DNA that contain genes of interest.
Vectors
After a desired segment of DNA is cut away from the donor’s genome with restriction endonucleases, it is ligatedinto a vectorDNA molecule, usual-ly a plasmid or a bacteriophage genome.Ligasesare enzymes that catalyze the formation of a phosphodiester bond between the 3 -hydroxyl group of a segment of donor DNA and the 5 -phosphate group of the vector DNA.
A vector is a DNA molecule into which foreign DNA moleules are ligated and inserted into cells so that the recombinant DNA can be replicated. Plas-mid vectors must also contain a marker, such as an antibiotic resistance gene, to facilitate the selection of bacterial cells that contain the plasmid. Vectors can be introduced into host cells by transformation or transduction (see Chapter 7). An expression vectoris a vector that carries a gene that can be efficiently transcribed and translated by the host cell.

Host Cells
A number of bacterial and yeast strains have been developed for recom-binant DNA experiments. In order for a given plasmid to be replicated by a host cell, the cell must recognize its origin of replication site (oriC). Recombinant plasmid vectors are normally introduced into competent cells by transformation and then selected using appropriate cell culture media. For example, if the vector contains an ampR gene that encodes resistance to ampicillin, the culture media would include that antibiotic to ensure that only transformed cells will grow. Another method for introducing recombinant DNA molecules into host cells is electroporation. In this method, a suspension of exponen-tially growing host cells is mixed with a solution of recombinant DNA molecules and exposed to a high electric field for a few milliseconds. The high voltage alters the structure of the membrane so that pores are tem-porarily formed, allowing plasmid DNA to enter the cell. This method is
fast and efficient.
If bacteria are used as the host to clone eukaryotic genes, certain steps must be taken to make it possible for the bacteria to make sensible mRNA and functional proteins, since bacteria do not possess mechanisms for processing eukaryotic pre-mRNA molecules. To do this, it is neces-sary to isolate already processed mRNA from the donor eukaryotic cells and convert the single-stranded RNA to double-stranded DNA. Reverse transcriptasefrom retroviruses (see Chapter 10) uses RNA templates for synthesizing DNA. The resulting DNA molecules, known as cDNA (for complementary DNA), can then be used for cloning in bacteria since they posses only intron-free protein-coding genetic information.
Note!
Common hosts are E. coli and S. cerevisiae.
Solved Problems
Solved Problem 8.1
How many fragments would be generated by a re-striction endonuclease in a plasmid that has two recognition sequences?
Because plasmids are circular molecules, two fragments would be generated. However, if the DNA were linear and contained two recogni-tion sequences, three fragments would be generated.
Solved Problem 8.2
Suppose the restriction endonuclease HindIII cuts a 6.0 kb linear piece of DNA into two fragments; an 800 bp fragment and a 5200 bp fragment. NarI cuts the DNA also into two fragments; frag-ments 1200 and 4800 bp long. Relative to the HindIII cut site, there are two possible ways in which NarI could have cut the DNA. How can you determine the correct cleavage site for NarI with respect to HindIII?
To determine this, one must subject the DNA to a double digest in which both the enzymes are allowed to cut the DNA at the same time. When the double digest is allowed to take place, if the three fragments that appear upon electrophoresis of the restricted DNA are 400, 800, and 4800 bp, then the only possible way in which the data can be interpreted is with the 1200 bp NarI fragment containing the HindIII recognition site
800 bp from the end of the linear piece of DNA. If the 4800 bp NarI frag-ment contained the cut site, you would visualize fragments of sizes 800, 1200, and 4000 bp after electrophoresing the doubly digested DNA.